编者按:
据统计,中国目前已有约 1500 家药物临床试验机构。仅 2024 年,全国就登记了 4900 项药物临床试验,其中 2500 余项为新药注册临床试验。每年有数十个创新药、上百个新药依据这些临床试验结果获批上市。[1]
然而,翻开这一单单金玉漂亮的成果,内里却是荒草败絮。
每收一个患者,入组费动辄上万;一个医生手里拿着几十上百个项目,但全程连患者的面都没见过;写病历、下医嘱、跟随访,明明需要专业的医学判断,干活的却是没有医疗资质的协调员。
丁香园调研了新药临床试验中的一线工作者,试图呈现一个更复杂的现实:在合规的框架之下,本应被严格遵循的规则,在现实中如何被理解、执行与变形。
本文作者:丁二丫
临床协调员李灵第一次直观感受到想象与现实之间的落差,是在一个高血压项目里。
那天,主任带来了一个考虑入组的患者,但第一次测量的时候,患者的血压低于入组标准。于是主任便和患者说,「你先去爬几层楼梯,活动活动,等会儿再回来量一次。」
站在一旁的李灵愣了一下。「连我都知道运动会影响血压,主任怎么可能不知道?想来,就是为了把患者收进来而已。」
几分钟后,李灵给气喘吁吁的患者重新测了一次血压,刚好达到入组要求。一个本来应该被排除的患者,就以这样神奇的方式进入了研究。
入组费几万一个人,主任:我的标准就是入组标准
像这样的事情,在新药临床试验中并不少见。某次项目中,张文接到协调员打来的电话。电话那头的人有些犹豫:「主任想让一个患者提前入组,但方案好像不允许。」
张文是一名临床监查员(CRA),职责是监督临床试验是否按照研究方案和规范执行。有涉嫌方案违背的事情,张文当然要介入。向协调员了解情况后,他很快意识到问题出在哪里。
那是一个肿瘤项目。患者之前一直在用其它抗肿瘤药物,按照方案,需要完成至少一周的洗脱期后,才能正式入组。可是当协调员把研究方案的要求告诉主任后,对方却有些不耐烦。
「怎么这么不懂变通,你别在病历上写他用过药不就行了?你这是在损害受试者的权益!」
但这种所谓的「灵活变通」,和研究规范是明显相悖的。
不论是什么类型的临床研究项目,对受试者都有较为严格的纳入和排除标准,通常包括年龄、疾病类型和分期、既往治疗史、身体状况及重要器官功能等。
将不符合这些要求的受试者收入研究中,不仅会让研究结果失真,更可能对入组的受试者造成损害。

某药物临床试验公示的入组标准(非文中提及项目) 图源:参考资料 5
李灵曾参与过的一个肿瘤项目,就差点发生这样的事情。
前期筛查时,研究者告诉项目组,这名患者整体状态不错,预期生存期也没有问题,强势要求把患者收入组。可真正见到患者时,李灵却发现情况并非如此。
患者坐在轮椅上,整个人瘦得厉害,几乎没有什么精神。问一句,停顿很久才慢慢回答一句。陪同前来的家属则显得格外疲惫。
沟通过程中,家属无意间提到,患者已经有一个多月没怎么吃东西了。
「我当时听到就觉得不对劲。」李灵回忆,「很多研究方案都会对受试者的整体状态作出要求,状态不佳的患者医疗风险太高,本来就不适合作为药物受试者。」
在她看来,这样的患者即便勉强进入研究,也未必能够完成后续访视和治疗,更难以承受试验药物可能带来的不良反应。
「这种事不是一回两回了。为啥?为了入组费呗。」
入组费,本质上是研究经费中的正常支出,用于补偿研究中心在招募、管理受试者方面的成本。入组费本身并不是灰色收入,但在一些项目中,高额的入组奖励也会悄悄改变规则——符合标准的患者不够,就想办法把不符合标准的人收进来。

图源:视觉中国
「比如一些肿瘤的项目,每入一个受试者,研究者能拿到五位数的入组费。」李灵表示,「在这种情况下,主任能想出一万种办法把患者收进来。」
协调员王升遇到过最离谱的一次,发生在一个医疗器械项目中。
那天,他在整理一个考虑入组的患者的检查报告,发现这个患者心脏超声报告上写的射血分数只有 25%。「这个数值明显不符合入组标准。」王升说。
按正常流程,这样的受试者应当被判定为筛查失败。可主任看完报告后,并没有讨论患者是否应该出组,而是当场拿起电话,打给了超声室。
「他就当着我的面,要求超声医生给患者复查一遍,并且把报告里的数值从 25% 改成 60%。」
手里握着几十个项目,却连受试者的面都没见过
对于一些研究者来说,「创收」的关键并不在于单个项目里能收多少患者,而在于自己手里能有多少项目。
虽然我国临床试验中心数量众多,但项目却高度集中在头部中心。在这些中心,一个头衔响亮的研究者手里常常同时管理几十个临床试验项目。
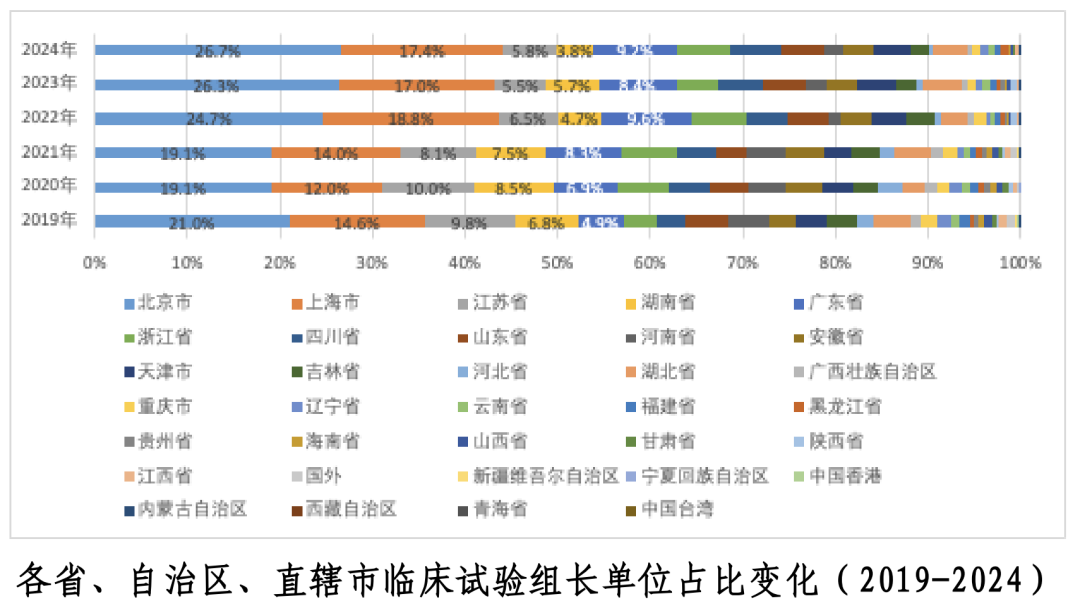
2024 年药物临床试验组长单位主要集中在北京、上海[1]
这些人大多是大科室的主任,临床工作本就繁重,再加上如此多的项目,时间和精力一分配,项目往往就变成了挂名之作,根本没人真正出力。
协调员吴锐记得,有一次一位受试者来院访视。按照规范,这本应是研究者评估病情、开具医嘱、记录病历并完成相关检查的一天。
之前给研究者发消息约访视时间,对方就一直没有回复。吴锐知道,他今天大概率也不会出现。
「事实上,这个研究者在整个试验期间基本就没怎么见过受试者。」吴锐无奈地说,「咱也不知道人家是真忙,还是纯不把临床试验规范当回事。」
于是,穿上白大褂的协调员坐到了电脑前。
先打开系统录入医嘱,再整理患者本次访视需要完成的检查项目;随后带着患者跑心电图、抽血、测量血压和血糖;等患者检查结束回来,又开始写病历。等忙完一圈,半天已经过去。
「连我整理好的资料,不到非签字不可时候,研究者都不会来签字。」
这样的场景,在临床试验的一线并不少见。写病历、开医嘱、做检查,「我就没听说过哪个协调员不用干这种活儿的。」不少协调员都这样说。
按照研究规范,开医嘱、书写病历、解读检查结果、进行随访和做医学判断等工作,都应由具备执业资格的研究者完成。临床协调员则仅是「在研究中心协助研究者完成大量非医学判断性工作的执行人员」,既没有做医疗判断的权限,也不具备开展临床操作的资质。

图源:社交平台
「如果要说完全合规的话,我的工作就只有安排访视、准备文件、提醒检查时间、录入数据、协调科室等等。」吴锐表示,「但我工作的绝大多数时间,都在干医生、护士该干的活儿。有同行说,每天上班打开待办事项,全都是职业红线。」
尽管研究者通常只是将与项目相关的医学事项交给协调员,但即便是这些,也已是对受试者的极度不负责。
一个临床研究圈子里都听说过的案例:在一家头部临床研究中心,一名协调员用研究者的账号登陆系统发药,将 4mg 的地塞米松误写成 40mg,造成了严重的后果。
「这个事情,确实一直都是传言,也没有什么官方通报。」有协调员表示,「但就算不是这个事,也肯定有类似的事。有的研究员只管拿钱签字,其它事情根本不管。」
从执行到监查,谁都难辞其咎
「有时候,我都觉得这些名头响当当的主任,无知得可笑。」
吴锐曾跟过一个多中心竞争入组的项目。这类项目的总入组人数是固定的,同时各研究中心也有各自的合同入组数。各中心同步招募,等整个项目达到总入组上限后,研究便会停止入组。
项目进行到后期时,总入组人数已经达到上限,申办方发来通知说,研究入组结束。在吴锐看来,这意味着所有中心都该停下来了。
可没想到,通知发出去后不久,主任却把她叫进办公室:「我们中心还有患者要进组。」
吴锐愣了一下,又翻出通知和主任确认。
「我不管,」主任摆摆手,「我们中心合同里的入组数还没完成,我就要入。」

图源:视觉中国
另一次,吴锐遇上了一个初次当研究者的主任。项目启动会上,申办方详细地介绍了研究方案,其中有一项是,受试者将按照随机原则分配至试验药组和安慰剂组,比例为 1:1。
散会后,主任把吴锐叫住,问她「这个比例能不能改一改?」
「啊?」吴锐以为自己听错了。
主任接着解释,希望自己中心入组的患者尽量都能分到试验药,最好不要分到安慰剂组。「这样对患者不是更好吗?」
「我都不知道说啥好了。」吴锐说,「有的主任脑子里仿佛根本没有『研究规范』这回事。」
当然,会出现这样离谱的事情,背后必然有更离谱的原因。
作为一种受监管的科学研究活动,新药临床试验原本有一套极其严格的伦理和操作规范,即《药品临床试验管理规范(GCP)》。根据规定,凡参与临床试验并承担职责的人,包括研究者、临床试验机构工作人员和临床研究协调员等,都应接受 GCP 培训,并在掌握规范后方可参与临床试验相关工作。[2]
但实际上,GCP 的培训和考试都并不严格,全程都在线上进行。吴锐透露,「很多主任的 GCP,都是协调员代考的,这都已经不是秘密了。」
一个新药的临床研究,通常由药企作为申办方,对项目进行设计、管理和监查,而临床试验中心(通常是医院)和临床试验现场管理组织(SMO)则是项目的执行方。
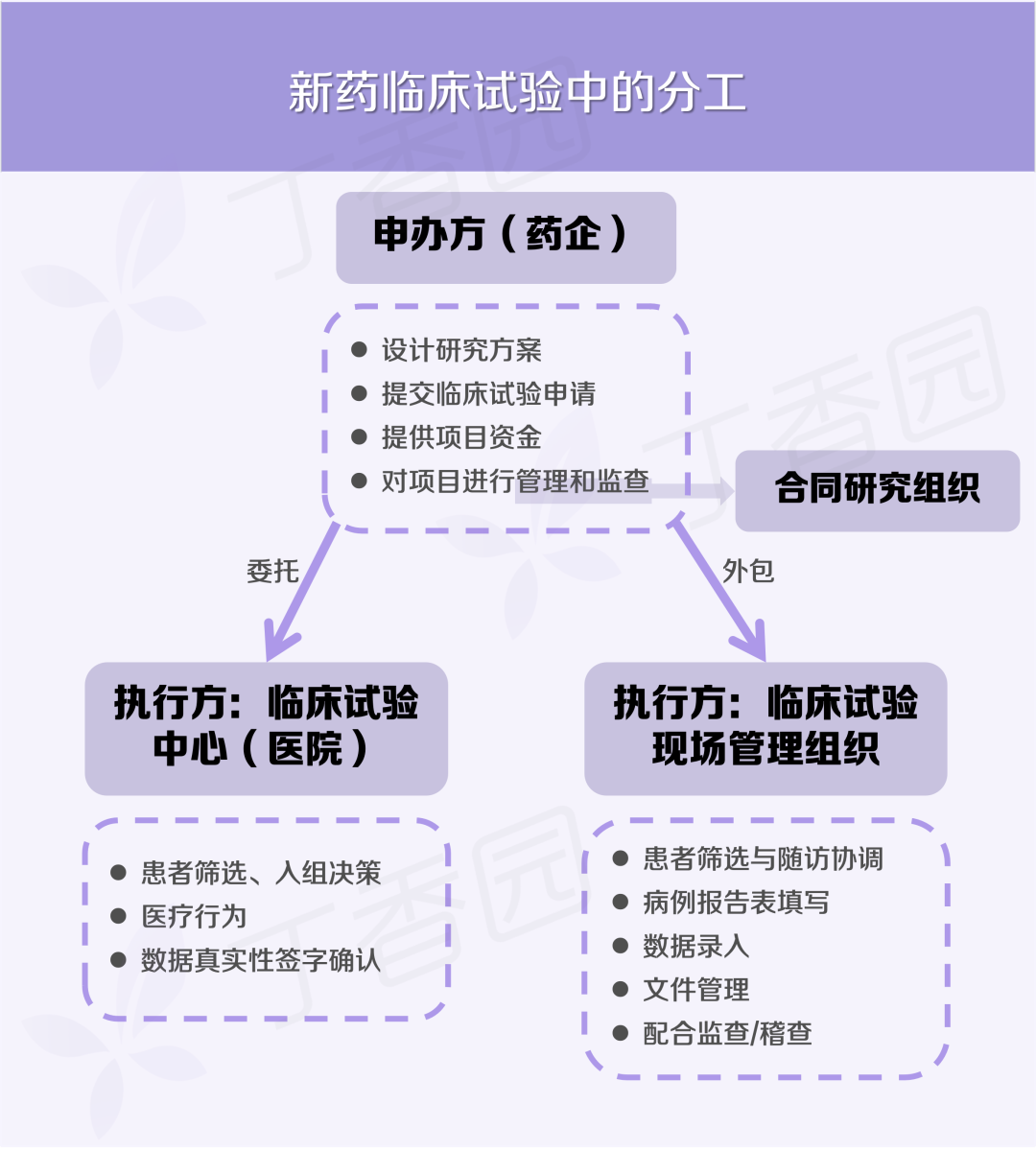
执行侧的乱象能够野蛮生长,监查侧的放任也难辞其咎。
协调员王升跟的那个项目,研究者让心超室把受试者的射血分数强行改为 60%,王升觉得离谱,就按流程上报了。
「但机构说,管不了这个研究者,事情就这么算了。最后这个患者就这么神奇地入组了。」
像这样「管不了研究者」的情况并不少见。虽然申办方是为研究提供资金的一方,但有时贪图一些知名的研究者在业内的影响力,就会对他们的行为睁一只眼闭一只眼。
「有的申办方只在乎结果,甚至会自己提出让协调员做做数据、瞒报严重不良事件。」李灵表示。
图源:社交平台
但并不是每个申办方都如此无视合规性的。张文所在的申办方,对项目操作的标准化管理就很看重。「有研究者想隐瞒受试者用药史,我们的项目经理知道后非常坚定地拒绝了,还说,像这样不重视 GCP 的研究者,宁愿以后不要入她的受试者,也不能违反原则。」
有业内人士表示,一些大型药企非常重视试验合规性,就会减少项目外包,亲自选聘人员来负责项目的管理和监查。但更多的企业,会将具体的项目管理和监查外包给合同研究组织(CRO)。
当然,外包给 CRO 本身并不违规,但问题是,一些 CRO 在项目中未必会秉持对研究负责的态度,甚至会进一步把监查工作外包给兼职的临床监查员。当一项工作被层层外包,质量把控就更加困难。
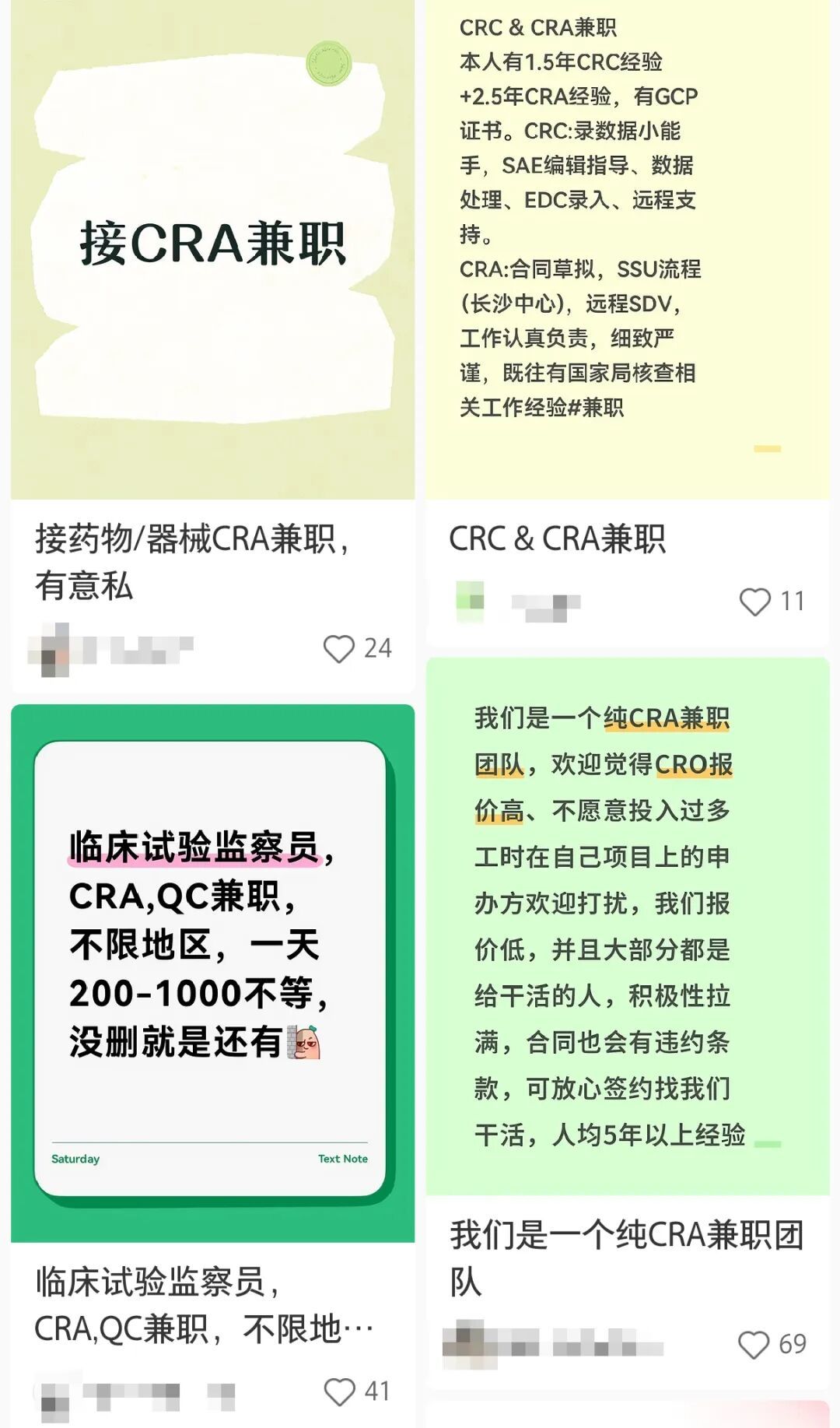
社交平台上的兼职临床监查员(CRA)
在国内药物临床试验的历史上,曾有过一次重要的监管风暴。
2015 年 7 月 22 日,原国家食品药品监督管理总局发布《关于开展药物临床试验数据自查核查的公告》,严厉整治临床试验数据造假,业内称为「722 事件」。[3]当年,食药监局要求 1622 个临床试验项目自查,最终超过 80% 的项目主动撤回了注册申请。[4]
「722 事件」后,临床试验的行业乱象得到了一定的整治。但十年多的时间过去,在烧过的土壤里,种子又生出藤蔓,结出一个个美丽但腐烂的果子。
「你别看过程怎么样,反正最终纸面上的东西,都漂亮得很。」
李灵、张文、吴锐、王升均为化名
策划:丁二丫|监制:islay
题图来源:视觉中国
参考资料
[1]https://www.cde.org.cn/main/news/viewInfoCommon/d0bc4836cfc4cb7c9ecf29ddaa7be6ea
[2]https://www.gov.cn/zhengce/zhengceku/2020-04/28/content_5507145.htm
[3]https://www.cde.org.cn/main/policy/view/ff096416e30a3256cf9c94ec38969954
[4]https://finance.people.com.cn/n1/2016/0621/c1004-28464196.html
[5]https://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml
特别声明:以上文章内容仅代表作者观点,不代表本站观点或立场。如有关于作品内容、版权或其它问题请于作品发表后与我们联系。